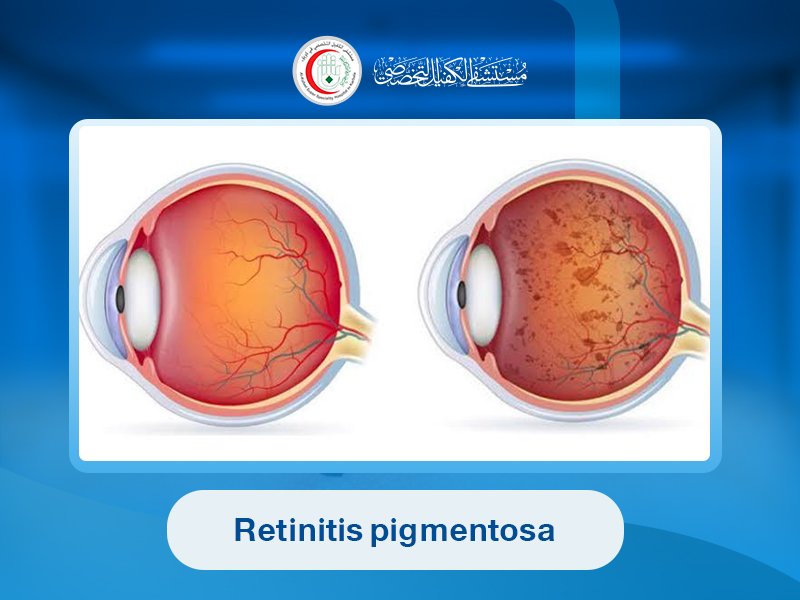
is the name given more than a century ago to a condition in which pigmented spots were observed at the base of the eye using ophthalmoscopy.
It was originally believed to be an inflammatory disease, but during the 20th century it was discovered that the condition is genetic, caused by gene defects. It also became clear that RP is one of the hereditary eye diseases most likely to lead to blindness with age.
There are multiple gene mutations that can cause the disease, but only a small number (fewer than ten) have been fully identified to date.
Retinitis pigmentosa is a rare disease, with a prevalence of approximately 1 in 2,700 people in the United States.
The retina contains two types of light-sensitive cells: rods and cones. Rods are located mainly in the outer retina and are activated in low-light conditions.
Most forms of retinitis pigmentosa primarily affect rods first, leading to loss of night vision and peripheral (side) vision.
Cones are mainly located in the central retina and are responsible for color vision and fine detail. When cones are affected, patients gradually lose central vision and color perception.
________________________________________
Symptoms of Retinitis Pigmentosa
Early symptoms usually appear in early childhood. The main symptoms include:
1. Night blindness (loss of night vision)
Difficulty seeing in low light may begin as early as age five. Night blindness is a universal and defining symptom of the disease.
Patients cannot see in the dark, although daytime vision may remain normal initially. Over time, adaptation to darkness becomes slower.
People may bump into objects, have difficulty driving at night, or struggle to see in dark environments such as cinemas.
2. Gradual loss of peripheral vision
This is often described as “tunnel vision.” Patients may bump into objects because they cannot see things in their side vision.
3. Loss of central vision
Some patients also develop central vision problems, making detailed tasks such as reading or sewing difficult.
Loss of visual field is very common (reported in about 94% of patients), while central vision loss varies widely.
Some individuals retain central vision until older age, while others lose it as early as their 30s.
Retinal changes vary with disease progression. Early stages may show no obvious signs in the first decade of life. In the second decade, pale spots appear in the retina along with dark pigment deposits. Later, retinal degeneration occurs with characteristic bone-spicule pigmentation.
________________________________________
Causes and Risk Factors of Retinitis Pigmentosa
More than 60 genes are associated with this disease. It can be inherited in several patterns:
1. Autosomal recessive inheritance
Both parents carry one copy of the mutated gene without symptoms. If a child inherits both copies, the disease appears.
Each child has a 25% chance of being affected.
2. Autosomal dominant inheritance
Only one copy of the mutated gene is needed for the disease to appear.
Each child has a 50% chance of inheriting the condition.
3. X-linked inheritance
The gene is carried on the X chromosome. Mothers may be healthy carriers but pass the gene to their sons. Affected males usually develop severe symptoms.
Fathers do not pass the disease to their sons in this form.
________________________________________
Complications of Retinitis Pigmentosa
• Early development of cataracts
• Swelling of the retina (macular edema)
________________________________________
Diagnosis of Retinitis Pigmentosa
Doctors may diagnose the condition through eye examination and the following tests:
1. Ophthalmoscopy
Eye drops are used to dilate the pupil, allowing examination of the retina. Characteristic dark pigment spots may be visible.
2. Visual field test
The patient focuses on a central point while responding to lights appearing in peripheral vision. This creates a map of visual field loss.
3. Electroretinography (ERG)
Electrodes or special lenses measure how the retina responds to flashes of light.
4. Genetic testing
A DNA sample is analyzed to identify the specific genetic type of retinitis pigmentosa.
________________________________________
Treatment of Retinitis Pigmentosa
There is currently no definitive cure. Past experimental treatments (such as early surgical implants) have not proven effective.
1. Treatments that may slow progression
• Acetazolamide: may reduce swelling in the central retina (macular edema) and improve vision.
• Vitamin A therapy: high doses may slow progression, but excessive intake can be toxic and must be medically supervised.
• Sunglasses: reduce light sensitivity and protect the eyes from UV exposure.
• Retinal implants: may provide partial vision in advanced stages.
2. Treatments under research
Current research focuses on:
• Gene replacement therapy (introducing healthy gene copies)
• Stem cell transplantation into the retina
• Electronic retinal implants that convert light into signals for the optic nerve
These approaches show promising potential for future treatment.
________________________________________
Prevention of Retinitis Pigmentosa
Genetic counseling and genetic testing can help identify whether a child is at risk of developing the disease.
-
Celebration of Imam Ali’s Birth Anniversary
Medical Units for Arbaeen Pilgrimage
Celebration of the Prophet Muhammad's (PBUH) Birth Anniversary
Celebration of Lady Zainab's (peace be upon her) Birth Anniversary
Bringing Joy to Patients on the Occasion of Imam Ali’s (peace be upon him) Birth Anniversary
Alkafeel Hospital Celebrates the Birth Anniversary of Imam Hussein (peace be upon him)
Joy Fills Alkafeel Hospital on the Birth Anniversary of Abu Al-Fadl Al-Abbas (peace be upon him)
Al-Kafeel Hospital participates in Arbaeen pilgrimage
-
Job Fair Participation at Al-Kafeel University
First Al-Kafeel Symposium on Musculoskeletal and Spine Surgery
IT Forum in Dhi Qar
University of Al-Kafeel Scientific Conference
Baghdad International Fair
Alkafeel Hospital Participates in the Sixth International Medical Sciences Conference at Al-Kafeel University
Alkafeel Hospital Participates in Iraq’s First Medical Tourism Conference, Affirming Its Pioneering Role in Health Sector Development
-
Training Lecture on Hospital Hospitality Services
Civil Defense Training Program
Training Program for Sama Karbala Foundation Staff
Workshop for Administrative Development Academy Trainees
Training for Turkish Hospital Medical Staff
Cervical Cancer screening Workshop
Scientific workshop for anesthesia students
Nursing Workshop for Dhi-Qar Province
Workshop on Quality and International Accreditation Standards
Hand Hygiene Training Workshop
Laboratory Training for Sama Karbala Foundation Staff
Wound Care Training for University Students
Scientific Workshop on General Surgery
Scientific Lecture on European Society of Cardiology Guidelines
Surgical Suturing Workshop for Medical Students
Accounting System Workshop for Administrative Development Academy Students
Risk Management Training
Training on Electrocardiography and Cardiac Arrhythmia Management
Proper Handwashing Techniques Workshop
Educational Lecture on Benign Prostatic Hyperplasia
Interactive Lecture for Medical Students of Al-Ameed University
Training on Cardiopulmonary Resuscitation for Dental Students
Security Staff Training on Explosives Detection Device “ID”
Delegation of Biomedical Engineering Students from Al-Safwa University
Human Resources Workshop for Hospital Departments
Radiation Protection Training Course
Quality of Medical Services Presentation
Fire Safety Training Workshop
Scientific Workshop on "Patient Safety"
Research Development Initiatives
Medical Training Workshop for University Students
Accounting Department Workshop
First Aid Training Workshop
Awareness Lecture on Early Breast Cancer Detection for Al-Siddiqa Al-Tahira Center
Training Program for Karbala Health Directorate Staff
Crisis Management and Fire Safety Training for Staff
Emergency Nursing Response Workshop
Students of the Administrative Development Academy Meet to Discuss the "Healthy Hospital" Project at Alkafeel Hospital
Successful Workshop on Workplace Etiquette Held at Alkafeel Hospital
Alkafeel Hospital Enhances Patient Satisfaction Through Specialized Training Course
Alkafeel Hospital Organizes Training Workshop to Enhance Anesthesiologists' Competency
Successful Breast Cancer Awareness Lecture at Alkafeel Hospital
Successful Workshop on PET Scan Technology Held at Alkafeel Hospital
Alkafeel Hospital Enhances Pharmacy Students’ Practical Skills
Alkafeel Hospital Emphasizes the Importance of Early Detection of Breast Cancer
Director of Alkafeel Hospital Discusses Development Strategies with General Surgery Team
Alkafeel Hospital Administration Strengthens Collaboration with Neurosurgery Team
Parliamentary Committee Commends Services at Alkafeel Hospital
Successful Workshop on Safety Practices Concludes at Alkafeel Hospital
Alkafeel Hospital Hosts Specialized Workshop on “Rezūm” Technology for Treating Benign Prostatic Hyperplasia
Alkafeel Hospital Enhances Efficiency of Its Human Resources Department
Organizational Meeting to Clarify the 2025 Postgraduate Application Process
Al-Kafeel Hospital Trains Its Staff on Safety and Evacuation Procedures
Sheikh Salah Al-Karbalai Delivers Religious Lecture to Alkafeel Staff
Alkafeel Training Center Concludes a Successful Scientific Day in Dentistry
Civil Defense Training at Alkafeel Hospital
English Debate at the Nursing Department
Meeting between Al-Kafeel Hospital and Al-Zaki Hospital in Babylon
Al-Kafeel Hospital participates in a workshop on Mass-Care Nursing
-
Spinal Surgery Patient Referrals
Spanish University Delegation Visit
Visit by Iraqi Ministry of Interior Delegation
Collaboration with Anwar Sheikha Hospital
Visit by College of Medicine, University of Diyala
Visit by Iraqi Ministry of Health Delegation
Shiraz University Delegation Visit
Visit by Faculty of Science – University of Karbala
Treatment of Pediatric Cardiac Patients
Memorandum of Understanding with Aster Medical Group
Delegation from Shahid Rajaei Hospital, Iran
Dr. Jassim Al-Ibrahimi Meets with Ministry of Health Referral Committee
Delegation of Medical Devices Students from Al-Mustaqbal University
Delegation of Medical Physics Students from University of Karbala
Student Delegation from Warith University
Meeting Between Dr. Jassim Al-Ibrahimi (CEO) and Department Heads
Visit by Croatian Academic Delegation
Meeting Between Al-Kafeel and Al-Battat Hospitals
Visit by Ministry of Health Delegation
Admission of Spinal Surgery Patients
Pediatric Cardiac Surgery Referrals
Meeting with Medical Students – Al-Ameed University
Collaboration with Karbala Nursing Syndicate
Visit by Al-Nahrain University Students
Dr. Jassim Al-Ibrahimi (CEO) Meets Top English Language Graduates
Visit by Al-Mustaqbal University Students
Alkafeel Hospital Holds Meeting with Dr. Moataz, Specialist in Internal Medicine
Alkafeel Hospital and Ministry of Water Resources Collaborate to Promote Public Health
High-Level Georgian Delegation Visits Alkafeel Hospital
French Delegation Visits Alkafeel Hospital: Toward Advanced Medical Collaboration
"A Leading Partnership to Support the Future of Engineers": Alkafeel Hospital Signs Cooperation Agreement with the College of Engineering at the University of Karbala
Alkafeel Hospital Provides Medical Care to the Blind Football Federation Players
High-Level Swiss Delegation Visits Alkafeel Hospital
Al-Nahrain University and Alkafeel Hospital Discuss Prospects for Joint Cooperation
President of the University of Babylon Explores Alkafeel Hospital’s Services and Discusses Future Cooperation
Alkafeel Hospital Commends the Civil Defense Directorate for Their Support
High-Level Georgian Delegation Visits Alkafeel Hospital
Academic Collaboration Between the University of Karbala and Alkafeel Hospital
Prospective Academic Cooperation Between the University of Karbala and Alkafeel Hospital
Ministry of Health Delegation Praises Dental Center at Alkafeel Hospital
Ongoing Collaboration Between Alkafeel Hospital and the Ministry of Health in Corneal Transplantation
Alkafeel Hospital Receives a New Group of Spinal Deformity Patients
Academic Delegation Praises Alkafeel Hospital
Expanding Health Insurance Coverage: Discussions Between the Al-Abbas Shrine and the Ministry of Health
Delegation from the Imamia Medics International Visits Alkafeel Hospital
Productive Academic Collaboration Between Al-Kafeel Hospital and the University of Babylon
College of Tourism at Al-Mustansiriya University: Al-Kafeel Hospital Sets a Distinguished Standard in Hospitality Services
Al-Kafeel Hospital Hosts Medical Delegation from Al-Muthanna Hospital
Citizens: Alkafeel is a Pioneer in Pediatric Cardiac Surgery and an Alternative to Medical Travel Abroad
Alkafeel Hospital Opens New Avenues of Collaboration with Maysan Health Directorate
Algerian Delegation Explores Alkafeel Hospital’s Advanced Medical Technologies
Qatari Delegation Visits Alkafeel Hospital to Discuss Joint Collaboration
Alkafeel Hospital Launches a Successful Vision Care Initiative in Maysan
Signing of a Cooperation Agreement between Alkafeel Hospital and the University of Karbala
Visit of Al-Ameed University Students to Alkafeel Hospital
Eye Examinations and Treatment for Students from Maysan Schools at Alkafeel Hospital
Visit of the Health Insurance Authority Delegation to Alkafeel Hospital
Alkafeel Hospital and Marina Al-Ahly Hospital Sign Memorandum of Understanding
Visit of Dr. Jassim Al-Ebrahimi to Marina Surgical Hospital
Al-Kafeel Hospital prepares for the Health Insurance Program
Link copied